At the electronic scale, first-principles methods such as Density Functional Theory (DFT) and Quantum Monte Carlo (QMC) leverage quantum mechanics to obtain insights into a material’s electronic structure. These models can elucidate bond characteristics, optimize lattice parameters, assess the relative thermodynamic stabilities of crystal structures, and analyze elementary excitations underlying all material properties. The length scale considered in these methods typically ranges on the order of nanometers, while the associated time scale operates at the order of picoseconds.
Stepping above the electron scale, molecular dynamics (MD) abstracts from the internal structure of atoms, portraying the system as atomic particles subject to Newtonian mechanics. MD has the capacity to simulate trajectories for millions of atoms, with ordinary MD reaching timescales on the order of nanoseconds and accelerated MD extending to microseconds. MD provides dynamic details about atomic-level system evolution, facilitating the comprehension of phenomena like nucleation in thin-film growth or defect formation and coalescence. Rare-event methods like Monte Carlo (MC) focus on events such as adsorption, desorption, or diffusion, disregarding vibrational motion and enabling MC simulations to span larger length and time scales, up to millimeters and seconds.
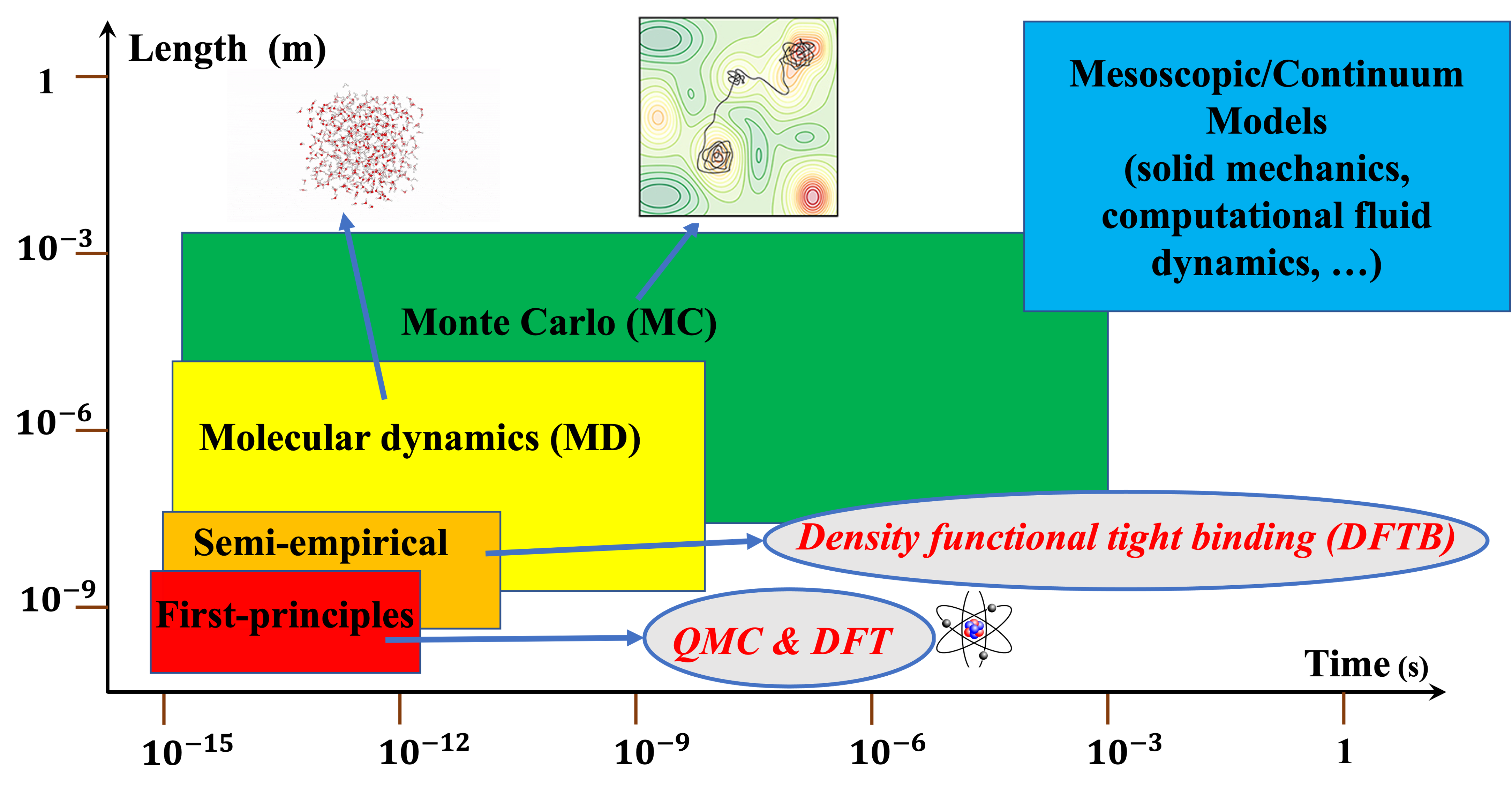
A higher resolution model necessitates increased information processing and becomes computationally demanding for larger time or length scales. A multiscale approach functions as a hierarchical structure where the outputs of a finer model become inputs for the next larger-scale model. Multiscale modeling aims to strike a balance, ensuring computational feasibility while mitigating the limitations of coarse-grained approximations by incorporating results from more detailed models.
The challenge lies in determining how information from finer models integrates into the coarse-grained model. In our group, we employ multiscale computational approaches relying on first-principles methods such as DFT and QMC to comprehend the intricate physical and chemical details of the synthesis process at the nanoscale. Subsequently, rather than relying on parametrization, we utilize machine learning interatomic potentials (ML-IAPs) to bridge the quantum-mechanical accuracy of QMC/DFT, in a nonparametric form, to larger scale simulations involving MD and MC.
STEAM Lab employs multiscale modeling leveraging ML-IAPs to simulate the large-scale synthesis of 2D materials with near-quantum accuracy, achieving significantly reduced computational costs. We investigate both top-down synthesis methods, such as chemical exfoliation, and bottom-up growth methods, such as chemical vapor deposition (CVD). Modeling synthesis processes at large scales with chemical accuracy aids in unraveling the complex underlying mechanisms governing these processes. This, in turn, accelerates the commercial production of 2D materials at wafer scale, expediting their integration into next-generation 2D devices.